
by Bhavika Sewpal
When the Human Genome Project determined the nucleotide sequence of every gene, it simultaneously revealed the amino acid sequence of every protein. That achievement was remarkable but also insufficient, because the linear chain of amino acids folds into a 3-dimensional (3D) structure, and knowing that structure is essential for determining the function of a protein. Indeed, for nearly a century, predicting 3D structure from linear structure has been a holy grail of biology. But with the advent of AI and machine learning, scientists have been able to produce sophisticated algorithms, enabling them to tackle this problem from a different angle. The most recent and notable one is the AlphaFold software, which has produced unprecedented results in the protein structure prediction field. But before delving into more complex topics, let’s make sure you understand all the basic terminologies first.
Table of Contents
Protein Structure
In simple terms, proteins are the basic building blocks of biological organisms. They are responsible for a wide array of processes that are vital in living systems. It is estimated that the human body contains between 80 000 and 400 000 proteins, which are essential for the proper functioning of different processes. For instance, the protein haemoglobin binds to oxygen molecules in the lungs and carries it to all the tissues in the body while the protein keratin plays an important structural role in our hair and nails. In addition, proteins form the backbone of our immune system and are responsible for keeping us healthy. Our body is able to fight against pathogens by secreting antibodies, which are a special type of protein. All these proteins play widely different roles in our bodies and this is possible because they all have different structures, which leads us to a very important question. What determines protein structure?
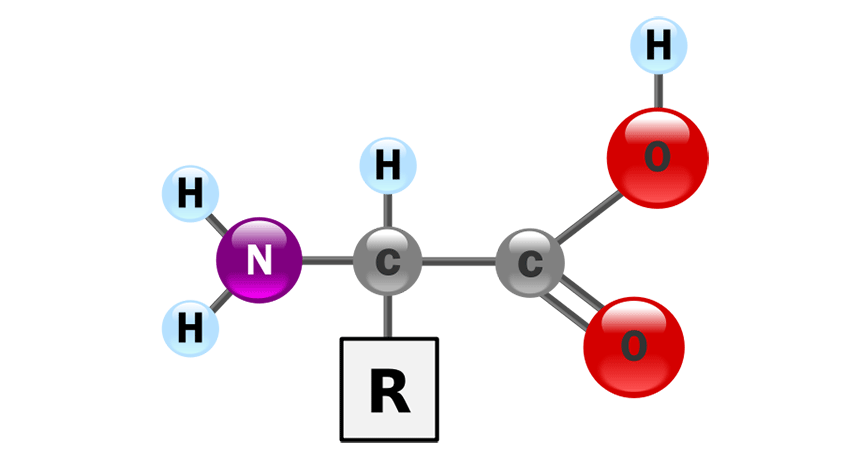
First, proteins are complex molecules, which are derived from much simpler molecules. The building blocks of proteins are amino acids. All amino acids have the above structure; they all have an NH2 group and a COOH group. The only place where they differ is in the R group.
Roughly 500 amino acids have been identified in nature but interestingly, only 20 are needed to make all the different types of proteins found in the human body. This means that the 80 000 to 400 00 proteins found within the human body can be synthesized by creating different sequences (or combinations) of various lengths of amino acids (all of which come from those 20 amino acids). This linear sequence of amino acids is known as the polypeptide chain and forms the primary structure of protein molecules.
There are 4 levels of protein structure. The first one is the primary structure as described above, and the others are the secondary, tertiary and quaternary structures. These subsequent structures mainly arise because of the various interactions between the R groups of the amino acids in the primary structure, which causes the linear polypeptide chain to fold into a specific 3-Dimensional shape.
The importance of predicting protein structures
Knowledge of the structure of a protein is the basic prerequisite for understanding its function. Currently, the main techniques used to determine protein structure are X-ray crystallography and nuclear magnetic resonance (NMR). In X-ray crystallography the protein is crystallized and then using X-ray diffraction, the structure of the protein is determined. However, this method is not always straightforward and can be very time-consuming, taking up to five years in some cases. NMR is also a useful technique for determining protein structure and offers the advantage that the protein can be studied in an aqueous environment, which may resemble its actual physiological state more closely [1]. However, one major disadvantage of NMR is that it is unsuitable for studying proteins that have more than 150 amino acids in their polypeptide chains, which greatly hinders advances in protein structure prediction. From an experimental point of view, the challenges associated with these two techniques are cost, time and expertise. To be able to use NMR and X-ray crystallography, a high level of specialization is needed and the cost of solving a new structure can reach up to $ 100 000.
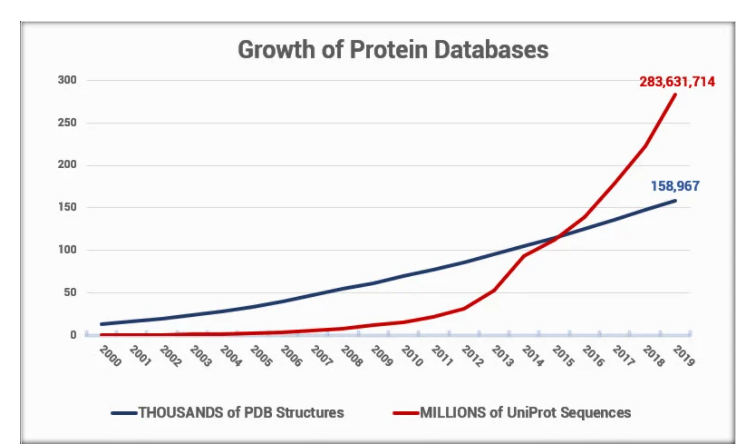
Due to a lack of efficient techniques in the protein structure prediction field, the gap between known protein sequences and the known protein structure is increasing exponentially. This can be illustrated in the graph below. The UniProt database, which contains information on protein sequences, is 1700 times bigger than the Protein Data Bank (PDB database), which stores known protein structures [2].
Practical uses of protein structure prediction
There is a lot of interest in structure prediction as a screening process for proteins that are not tenable for experimental determination. For instance, companies specializing on antibody development can generate thousands of antibody sequences in response to targets. As a result, there is a need to screen out antibodies that do not have the desired characteristics as quickly as possible. One way to improve this quick-to-fail mentality is to use structure prediction to predict properties of a protein fold based on the amino acid sequence [2].
If the proper techniques are developed, in the future, scientists might be able to develop enzymes (which are highly specialized proteins) that will be able to digest plastics and other industrial waste, which will no doubt be a valuable tool to combat pollution. In short, a breakthrough in the field of protein prediction will have widespread positive impacts in various spheres of our lives.
The Protein Folding Problem and Levinthal’s Paradox
In his acceptance speech for the 1972 Nobel Prize in Chemistry, Christian Anfinsen famously postulated that, in theory, a protein’s amino acid sequence should fully determine its structure [3]. This hypothesis sparked a five decade quest to be able to computationally predict a protein’s 3D structure based solely on its 1D amino acid sequence as an alternative to expensive and time consuming experimental methods such as X-ray crystallography and NMR .
A major challenge, however, is that the number of ways a protein could theoretically fold before settling into its final 3D structure is astronomical. In 1969 Cyrus Levinthal noted that it would take longer than the age of the known universe to enumerate all possible configurations of a typical protein by brute force calculation – Levinthal estimated 10^300 conformations for a typical protein. Yet in nature, proteins fold spontaneously, some within milliseconds – a dichotomy sometimes referred to as Levinthal’s paradox.
CASP
Critical Assessment of Protein Structure Prediction (CASP) is one of the organizations that has heavily contributed to drive progress in the protein structure prediction field. Essentially, CASP is a community-wide, worldwide experiment for protein structure prediction and has been taking place every 2 years since 1994. The competition’s format is simple. Contestants are given the one-dimensional amino acid sequences for roughly 100 proteins whose three-dimensional structures have been experimentally determined but are not yet publicly available. Using the amino acid sequences as inputs, the competitors generate predictions for the proteins’ structures, which are then compared against the “ground truth” structures to determine their accuracy.
The primary method of evaluation is the Global Distance Test (GDT), also written as GDT_TS (to represent total score). GDT_TS is a measure of similarity between two protein structures with known amino acid correspondences (for instance, identical amino acid sequences) but different tertiary structures (i.e. different folding). It is most commonly used to compare the results of protein structure prediction to the experimentally determined structure as measured by X-ray crystallography, NMR or, increasingly, cryoelectron microscopy. This metric is intended as a more accurate measurement than the common root-mean-square deviation (RMSD) metric – which is sensitive to outlier regions created, for example, by poor modeling of individual loop regions in a protein structure that is otherwise reasonably accurate. The conventional GDT_TS score is computed over the alpha carbon atoms and is reported as a percentage, ranging from 0 to 100. In general, the higher the GDT_TS score, the more closely a model approximates a given reference structure [5].
Calculation of the GDT score
The GDT score is calculated as the largest set of amino acid residues’ alpha carbon atoms in the model structure falling within a defined distance cutoff of their position in the experimental structure, after iteratively superimposing the two structures. By the original design, the GDT algorithm calculates 20 GDT scores, i.e. for each of 20 consecutive distance cutoffs (0.5Å, 1.0Å, 1.5Å, … , 10.0Å). For structure similarity assessment, GDT scores from several cutoff distances are used, and scores generally increase with increasing cutoff. A plateau in this increase may indicate an extreme divergence between the experimental and predicted structures, such that no additional atoms are included in any cutoff of a reasonable distance. The conventional GDT_TS total score in CASP is the average result of cutoffs at 1, 2, 4, and 8.
The GDT_TS is commonly interpreted as an approximation of the area under the GDT curve, denoted by GDT_A. Unfortunately, since the measure is approximated using the GDT function values at only several distance cutoffs, the errors in the area approximation are large. Nevertheless, the graph of a GDT function provides valuable insight into the quality of a protein model. More specifically, the closer the graph runs to the horizontal axis (in other words, the smaller the area under the graph), the better the model [4].
DeepMind’s AlphaFold triumph in CASP 14
AlphaFold’s performance at last year’s CASP was historic, far eclipsing any other method to solve the protein folding problem that humans have ever attempted. On average, DeepMind’s AI system successfully predicted proteins’ three-dimensional shapes to within the width of about one atom. The CASP organizers themselves declared that the protein folding problem had been solved. Before AlphaFold, the 3D structures for only about 17% of the roughly 20,000 proteins in the human body was known [6]. Those protein structures that were known had been painstakingly worked out in laboratories over decades through tedious experimental methods like X-ray crystallography and NMR. By using the AlphaFold algorithm, the 3D structures for virtually all (98.5%) of the human proteome are now known. Of these, 36% are predicted with very high accuracy and another 22% are predicted with high accuracy. [7]
AlphaFold
Essentially, AlphaFold solves the problem in two steps. The first step involves a deep learning predictive modeling (convolutional neural network), and the second step involves applying the gradient descent optimization.
Like any other machine learning model, the solution started with the training data. AlphaFold was trained on a few different publicly available datasets for instance the Protein Data Bank (PDB) and the UniProt database. In this case, the input were the protein sequences (a sequence of amino acids). Before doing any kind of prediction however, a feature engineering algorithm needed to be applied to the input. The technique that AlphaFold used is called MSA (multiple sequence alignment).
To give a basic intuition, imagine we have a long chain of amino acids, and parts of this chain are similar to some parts of other known proteins. Using alignment, we can identify those similar subsets and make a feature profile for that section of our sequence. The AlphaFold team did this kind of feature engineering for 31,247 proteins from Protein Data Bank (PDB) and split them into the train and test sets (29,427 and 1,820 proteins, respectively) [8]. In addition, they calculated the distance and torsion angles between each pair of protein residues (i.e. amino acids). This would in fact be the target for the predictive model. Now that they had the MSA features (as input) and distogram matrices (matrix of distances between different parts of a protein) as a target, they needed a model to train and test them. For this purpose DeepMind team decided to use a Deep Res-Net model (a deep convolutional neural network model).
So, step one outputs two matrices that show the distance and torsion angles between different parts (or residues) of a protein. To obtain a 3D structure, these two matrices must be translated into a 3D structure. For example, in a protein sequence, the AlphaFold program should be able to determine where there is a bond or a kind of torsion. The process of converting the distogram matrices into 3D structures is done with an iterative gradient descent method [8]. To achieve this goal, the algorithm starts with a smooth 3D structure model, updates the structure until the distogram of the predicted structure gets as close as possible to the distogram (or output) from the deep learning part.
In a nutshell, by implementing neural networks, DeepMind’s AI was able to solve the computational problem of predicting protein structures from protein sequences. Several improvements were made in the neural networks of subsequent versions of AlphaFold in order to achieve higher GDT_TS scores in CASP 14. However, despite these tremendous successes, AlphaFold still has its limitations. As is the case with most machine learning models, the capacity of the model is somewhat restrained by the nature of the training data. So, AlphaFold tends to perform poorly on determining the structures of proteins which are radically different from those in the PDB database. Nonetheless, this remains a huge success for the computational biology field and we will surely reap the benefits of this new technology over the next few decades as all areas of biology benefit from this resource.